


On the significance of 21 USC 360bbb-3(k): "use" of EUA products "shall not constitute clinical investigation."

Last week I got an email requesting clarification about the significance of 21 USC 360bbb-3(k) for the planning, execution and continuance of the Covid-19 global pharmaweapon mass murder campaign.
21 USC 360bbb-3 Authorization for medical products for use in emergencies
…21 USC 360bbb-3(k) Relation to other provisions
If a product is the subject of an authorization under this section, the use of such product within the scope of the authorization shall not be considered to constitute a clinical investigation for purposes of section 355(i), 360b(j), or 360j(g) of this title or any other provision of this chapter or section 351 of the Public Health Service Act [42 U.S.C. 262].
My reply, revised/expanded
The shortest version is that — like the current Good Manufacturing Practice (cGMP), current Good Laboratory Practice (cGLP), current Good Distribution Practice (cGDP) and labeling and dispensing laws that Sasha Latypova has investigated so thoroughly (and found that none of the standards that FDA applied to drug, vaccine and biologics development prior to 2020, were applied by FDA to the products produced after the 2020 PREP Act declarations about Covid-19 EUA countermeasures) — so also none of the current Good Clinical Practices (cGCP) were followed either.
Brook Jackson identified these blatant violations in the human clinical "trials" in August and September 2020, collected supporting evidence, and described the violations in detail, with supporting documentation and photos, in her reports to Ventavia, Pfizer and FDA.
Ventavia, Pfizer and FDA ignored the evidence; continued attacking unwitting victims with lethal injections while telling those victims they were participants in an FDA-regulated clinical trial; and arranged for Jackson to be fired.
Jackson included the same information and evidence in her whistleblower complaint at p. 8
…[Brook Jackson] observed:
fabrication and falsification of blood draw information, vital signs, signatures and other essential clinical trial data;
enrollment and injection of ineligible clinical trial participants, including Ventavia employees’ family members;
failure to timely remove ineligible patients’ data from the trial;
failure to maintain temperature control for the vaccine at issue;
failure to monitor patients after injection as required by the trial protocol;
principal investigator oversight failures;
use of unqualified and untrained personnel as vaccinators and laboratory personnel;
failure to maintain the “blind” as required, which is essential to the credibility and validity of the observer-blinded clinical trial;
ethical violations, such as failure to secure informed consent and giving patients unapproved compensation;
improper injection of the vaccine (i.e., by over-diluting vaccine concentrate or using the wrong needle size);
failure to ensure that trial site staff were properly trained as required by good clinical practices;
safety and confidentiality issues, including HIPAA violations; and
other violations of the clinical trial protocol, FDA regulations, and Federal Acquisition Regulations and their DoD supplements.
Ventavia failed to report the majority of its clinical trial protocol and regulatory violations to Pfizer or the external Institutional Review Board. Issues were improperly documented or hidden away in “notes to the file,” and not corrected…
If any FDA regulations had been legally operative, then the whole project would have been stopped by FDA long before human sham-trials could even begin.
Red flag stopping points showed up in the very earliest animal studies, one of which was conducted between July 16, 2020 and Sept. 24, 2020, concurrent with the sham human trials, and eventually provided by Pfizer/Acuitas/DOD to FDA in November 2020.
Another version was provided to Japanese regulators by February 2021, after mass rollout worldwide began in December 2020. It was subsequently translated into English and discussed by Byram Bridle in May 2021 reports and on Bret Weinstein’s June 2021 Darkhorse podcast, highlighting that the data showed the lipid nanoparticles (payloads unidentified) accumulate in rat organs, among other toxicity evidence.
…At this point in early Summer 2021, four facts became more widely understood among the community of people trying to understand the biotechnology, risks and benefits of the products marketed as ‘Covid-19 vaccines.’
The inflammatory lipid nanoparticles and their payloads collect in the ovaries and other key organs, are not rapidly cleared from the human body and are toxic.
Pfizer scientists knew this before seeking EUA approval from the FDA through the 11/20/2020 EUA application.
FDA scientists led by Marion Gruber knew this when authorizing the product for emergency use on 12/11/2020.
Pfizer, FDA and Gruber withheld this information from the public and knowingly lied each time they described the products as “safe and effective…”
The Pfizer-DOD death machine submitted the Wistar rat data to the fake FDA reviewers as part of the EUA package, including a document called “Phase 1/2/3, placebo-controlled, randomized, observer-blind, dose-finding study to evaluate the safety, tolerability, immunogenicity and efficacy of SARS-CoV-2 RNA vaccine candidates against Covid-19 in healthy individuals.”
In that sham “clinical trial” protocol at p. 72, Pfizer-DOD flatly stated that the “study” had not and would not assess pharmacokinetics, pharmacodynamics, biomarkers or genetics.
The aggregate evidence for the intent and function of 21 USC 360bbb-3(k) as a blanket waiver of the American drug regulation system to facilitate and pre-cover-up a covert, criminal bioweapons production and deployment program — can be summed up as "the dog that didn't bark."
Reinforcing evidence is the establishment of "real world evidence" — “data regarding the usage, or the potential benefits or risks, of a drug derived from sources other than randomized clinical trials” — as a basis for fake FDA regulatory decisions, a monstrosity Congress passed and Obama signed through the 2016 21st Century Cures Act at Section 3022. More reinforcing evidence: the government-coordinated, fraud-based suppression of all the alternative treatments for Covid-19, any one of which would have been enough to block the EUA, which depends on there being no available alternative treatments.
Another way to think about 21 USC 360bbb-3(k):
It’s the provision that quietly nullified every substantive way in which FDA regulatory functions would have been fulfilled, rendering the entire FDA performance a sham intended only to shield from public view, that the operation was and is actually run under 50 USC Ch. 32, the Chemical and Biological Warfare Program.
As I keep researching, I find more evidence that FDA officials fully understood how outside-the-FDA-law the EUA program is, and they’ve understood it for a very long time.
Especially FDA lawyers running the “legal preparedness” apparatus.
See, for example, Susan Sherman's part in a 2009 workshop (Medical Countermeasures Dispensing Emergency Use Authorization and the Postal Model, at p. 26) and an August 2020 presentation by Elizabeth Sadove, summarizing the simultaneous cover-up/crime in a table at p. 18.
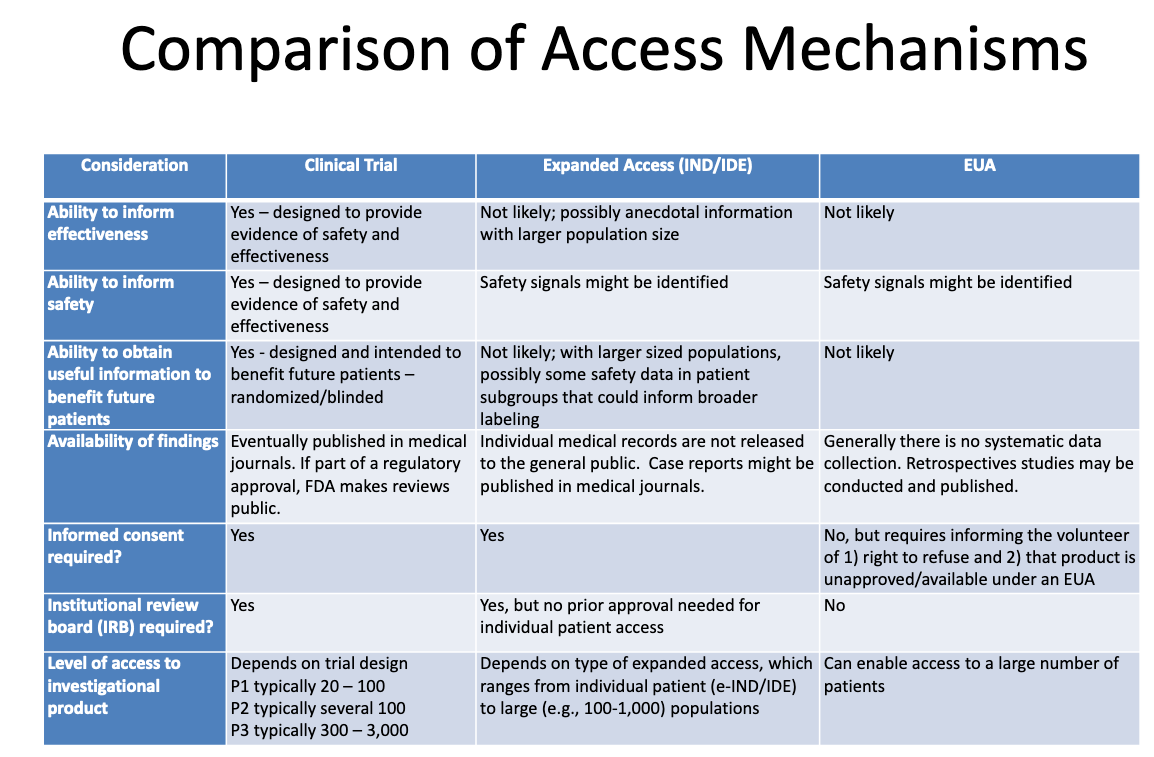
That table makes clear that "Clinical Trial" products, "Expanded Access (IND/IDE)" products and "EUA" products are three completely different legal frameworks.
Under "Clinical Trials," the use will provide evidence of safety and effectiveness; will produce useful information to benefit future patients; will eventually be published in medical journals and possibly published FDA reviews; that informed consent is required; that Institutional Review Boards are required; and that a limited number of people will have access to the product.
Under EUA, product use is "not likely" to provide evidence of efficacy; "might" provide safety signals; is "not likely" to provide useful information to benefit future patients; "generally there is no systematic data collection" although retrospective studies "may" be conducted and published; informed consent is not required; IRB review is not required; and the access pool is "a large number of patients."
The primary purpose of all the statutory, regulatory changes and guidance document revisions year after year, page after page, is to keep people from, first, understanding the war crimes as war crimes, and — if people do figure it out — keep them chasing their tails trying to find the FDA loophole that the war criminals somehow failed to close, through which somebody might someday be able to get them to stop killing us.
In the meantime, they just keep killing, and we don’t find loopholes, because the complexity of the web is impenetrable, and the program is not an FDA-regulated medical treatment program anyway: it’s a military-operated global genocide.
I try to maintain attention and expand understanding of demonstrable fact sets and the moral judgments that follow once those acts are accurately perceived:
"What they are doing is intentional killing, and intentionally killing people is wrong.”
And I try to participate in the global struggle to stop the killing by helping to mobilize political and social pressure on lawmakers, prosecutors and judges to use international and federal criminal laws to stop the cull and bring the killers to justice; repeal the enabling laws and put in place new laws that better protect people from socially- and economically- coerced submission to mass murderers pretending to be everything other than what they are.
Related:
April 25, 2022 - The investigational drugs that weren’t.

In politics, nothing happens by accident. If it happens, you can bet it was planned that way.
Franklin D. Roosevelt
Your work is appreciated. Keep it up, please.
Those of us who instinctively knew need no clarification- those who blindly follow, still will not accept the truth based on evidence or facts. It has been a well thought out plan of Crimes Against Humanity since the beginning. The number of those knowingly involved is staggering.
What I fail to understand is why people aren’t raising Sam-hell.